Progettazione & Engineering
Progettazione Meccanica
Rapid Prototyping
Reverse Engineering
Simulazioni FEM
Qualità e Validazione
Sistema Qualità
Validazione DQ IQ OQ PQ
Procedure Operative (SOP) (IOP)
Fascicolo Tecnico / Design Dossier
Design Control
DHF Design History File
Analisi e Gestione del Rischio
Valutazione Clinica
Testing
Test Meccanici
Analisi su Polimeri
Valutazione della Sicurezza Biologica
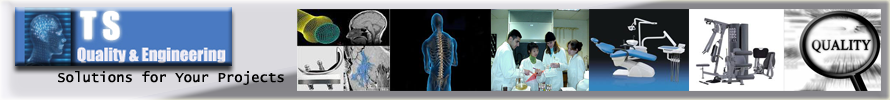
Siamo specializzati nell'ottimizzazione del processo di stampaggio ad iniezione.
Lo stampaggio a iniezione è un processo di produzione industriale in cui un materiale plastico viene fuso e iniettato ad elevata temperatura e pressione per consentire il riempimento di uno stampo chiuso, il quale viene aperto e rilascia il componente dopo il raffreddamento dello stesso.
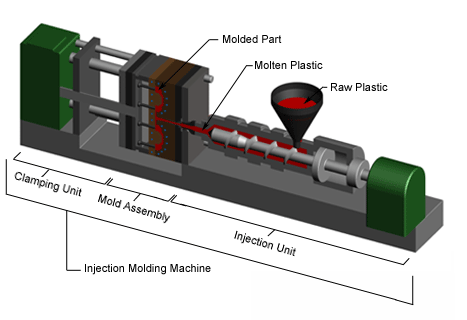

La realizzazione di un nuovo dispositivo viene condotta seguendo diverse fasi.
Siamo in grado di seguire lo sviluppo e la realizzazione di un nuovo dispositivo da produrre attraverso stampaggio ad iniezione in tutte le fasi.
Siamo specializzati nella progettazione e realizzazione di stampi per il settore medicale.
Validazione IQ OQ PQ
Progettazione & Engineering
Progettazione Meccanica
Rapid Prototyping
Reverse Engineering
Simulazioni FEM
Qualità e Validazione
Sistema Qualità
Validazione DQ IQ OQ PQ
Procedure Operative (SOP) (IOP)
Fascicolo Tecnico / Design Dossier
Design Control
DHF Design History File
Analisi e Gestione del Rischio
Valutazione Clinica
Testing
Test Meccanici
Analisi su Polimeri
Valutazione della Sicurezza Biologica
Eseguiamo convalide di processo con metodologia IQ OQ PQ in ottemperanza alle norme cGMP.
L'Annex 15 del EU GMP fornisce le linee guida per IQ OQ PQ convalida. Tutte le attività di convalida devono essere pianificate. Gli elementi chiave di un programma di convalida devono essere chiaramente definiti e documentati in un piano generale di convalida (VMP) o documenti equivalenti.
Il VMP deve essere un documento di sintesi, breve, conciso e chiaro. Il VMP deve contenere dati almeno i seguenti:
(a) validation policy;
(b) organisational structure of validation activities;
(c) summary of facilities, systems, equipment and processes to be validated;
(d) documentation format: the format to be used for protocols and reports;
(e) planning and scheduling;
(f) change control;
(g) reference to existing documents.
Lo scopo di un DQ validazione / IQ / OQ / PQ è quello di documentare e testare la composizione, il funzionamento e le prestazioni della macchina / processo o di prodotto in questione.
Il Protocollo di Validazione viene redatto secondo le linee guida GMP (Good Manufacturing Practice) e GAMP 4 (Good Automated Manufacturing Practice) nate ed utilizzate per il settore farmaceutico.

Attualmente anche il settore dei dispositivi medici e quello dei cosmetici stanno adottando tali linee guida al fine di ottenere uno standard qualitativo elevato delle macchine che realizzano, manipolano e confezionano prodotti rivolti ai suddetti settori merceologici.
Forniamo supporto nei seguenti ambiti:
- Convalida Processi di Sterilizzazione
- Convalida Processi di Lavaggio
- Convalida Processi Produzione Farmaceutica
- Convalida Trattamenti Superficiali
- Convalida Processo Stampaggio ad Iniezione
- Convalida Processi Speciali
Di seguito alcune definizioni atte a chiarire l'ambito applicativo delle diverse fasi di validazione:
Design Qualification
The first element of the validation of new facilities, systems or equipment could be design qualification (DQ). The compliance of the design with GMP should be demonstrated and documented.
Installation Qualification
Installation qualification (IQ) should be performed on new or modified facilities, systems and equipment. IQ should include, but not be limited to the following:
(a) installation of equipment, piping, services and instrumentation checked to
current engineering drawings and specifications;
(b) collection and collation of supplier operating and working instructions and
maintenance requirements;
(c) calibration requirements;
(d) verification of materials of construction.
Operational Qualification
Operational qualification (OQ) should follow Installation qualification. OQ should include, but not be limited to the following:
(a) tests that have been developed from knowledge of processes, systems and equipment;
(b) tests to include a condition or a set of conditions encompassing upper and lower operating limits, sometimes referred to as “worst case” conditions.
The completion of a successful Operational qualification should allow the finalisation of calibration, operating and cleaning procedures, operator training and preventative maintenance requirements. It should permit a formal "release" of the facilities, systems and equipment.
Performance Qualification
Performance qualification (PQ) should follow successful completion of Installation qualification and Operational qualification. PQ should include, but not be limited to the following:
(a) tests, using production materials, qualified substitutes or simulated product,
that have been developed from knowledge of the process and the facilities,
systems or equipment;6
(b) tests to include a condition or set of conditions encompassing upper and
lower operating limits.
Although PQ is described as a separate activity, it may in some cases be appropriate to perform it in conjunction with OQ.
Evidence should be available to support and verify the operating parameters and limits for the critical variables of the operating equipment. Additionally, the calibration, cleaning, reventative maintenance, operating procedures and operator training procedures and records should be documented.
Fascicoli tecnici
Progettazione & Engineering
Progettazione Meccanica
Rapid Prototyping
Reverse Engineering
Simulazioni FEM
Qualità e Validazione
Sistema Qualità
Validazione DQ IQ OQ PQ
Procedure Operative (SOP) (IOP)
Fascicolo Tecnico / Design Dossier
Design Control
DHF Design History File
Analisi e Gestione del Rischio
Valutazione Clinica
Testing
Test Meccanici
Analisi su Polimeri
Valutazione della Sicurezza Biologica
Il DHF è necessario secondo l' FDA per l'introduzione nel mercato degli Stati Uniti, e descrive un prodotto attraverso il suo ciclo di sviluppo, e secondo il Design Control, il suo ouput è il DMR (Device Master Record) che definisce il prodotto attualmente in commercio.
Tuttavia, l'UE (Unione Europea) ai sensi della direttiva sui dispositivi medici (MDD; Direttiva UE 93/42 / CEE) e per la marcatura CE, richiede un file che descriva il prodotto in un punto nel tempo, vale a dire, come attualmente commercializzato nell'UE / paesi del mercato comune, simili al DMR, con elementi del DHF, come documenti di gestione del rischio, e dati clinici.
La direttiva 93/42/CEE, specifica in dettaglio la documentazione tecnica che il fabbricante di un Dispositivo Medico D.M. deve realizzare per l'ottenimento della marcatura CE.

Generalmente il Technical File (TF) si rivolge ad un prodotto che è MDD di classe I e di classe IIa o IIb, mentre il Design Dossier (DD) riguarda un prodotto che è MDD Classe III.
Questi file sono composti dalle seguenti sezioni di base per sostenere la direttiva sui dispositivi medici (MDD), i requisiti essenziali (per questo prodotto), e la "Dichiarazione di conformità" emessa dal produttore:
- General Information / Product Description / EC Authorized Representative
- Classification Determination (Annex IX, Rule [select applicable rule])
- Essential Requirements (Annex I)
- Risk Analysis
- Labeling
- Product Specifications
- Design Control
- Clinical Evaluation (Annex X; literature review, et al.)
- System Test Reports
- —Functional Bench Testing
- —Lab Testing (cytotox, hemolysis, sensitization, carcinogenicity, other biocompatibility testing)
- —Sterilization validation (or AAMI TIR 28 Analysis)
- Packaging Qualifications
- Manufacturing
- Sterilization
- Conclusion
- Declaration of Conformity (Annex II, V, VII)
- Appendix (further supporting information / details on the above).
Newer EU regulatory guidance documents are moving the TF/DD more in the direction of the DHF.
Con il termine Fascicolo Tecnico si identifica una raccolta di tutta la parte documentale che ha coinvolto la progettazione, la costruzione, la validazione. In esso sono inclusi i calcoli strutturali, le caratteristiche dei materiali, i disegni di progetto, gli schemi, i criteri di progettazione, la valutazione clinica, l'analisi dei rischi.
Contrariamente al manuale di uso e manutenzione, il F.T. non deve essere fornito al cliente, bensì essere archiviato presso il costruttore.
Analisi dei Rischi
Progettazione & Engineering
Progettazione Meccanica
Rapid Prototyping
Reverse Engineering
Simulazioni FEM
Qualità e Validazione
Sistema Qualità
Validazione DQ IQ OQ PQ
Procedure Operative (SOP) (IOP)
Fascicolo Tecnico / Design Dossier
Design Control
DHF Design History File
Analisi e Gestione del Rischio
Valutazione Clinica
Testing
Test Meccanici
Analisi su Polimeri
Valutazione della Sicurezza Biologica
Per soddisfare le esigenze degli standard internazionali e garantire che la Vostra azienda introduca un prodotto sicuro, efficace nel mercato nei tempi e con i budget previsti, è necessaria la corretta implementazione del sistema di gestione del rischio. Possiamo assistere e aiutare la vostra azienda a farlo nel modo giusto.
Nel settore dei dispositivi medici, la gestione del rischio va oltre lo sviluppo e la produzione; è una parte vitale di tutti i processi della vostra azienda. La ISO 14971 definisce i requisiti internazionali dei sistemi di gestione dei rischi per i dispositivi medici, la definizione di migliori pratiche in tutta l'intero ciclo di vita di un dispositivo.
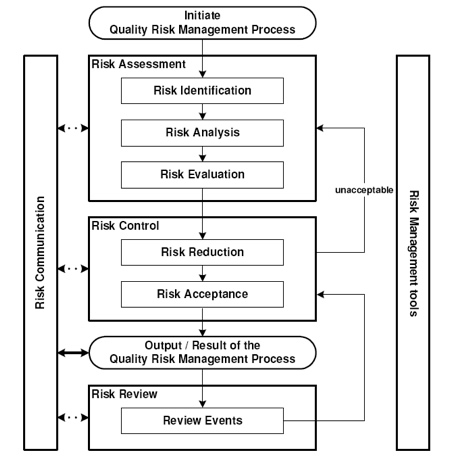
Panoramica di un processo di gestione dei rischi (fonte: GMP allegato 20)
Il produttore, a fronte di 93/42 CEE è tenuto ad effettuare un'analisi dei rischi al fine di evitare o ridurre al minimo la possibilità di incidenti.
ISO 14971: 2007 specifica un processo per un fabbricante di individuare i rischi connessi con i dispositivi medici, per stimare e valutare i rischi associati, di controllare questi rischi, e per monitorare l'efficacia dei controlli. A seguito di alcune definizioni importanti per la comprensione del processo di risk management:
|
Harm |
Damage to health of recipient, including the damage that can occur from loss of product quality or availability (loss of tissue, damage for the donor) |
|
Hazard |
The potential source of harm |
|
Risk assessment |
contains risk identification, risk analysis and risk evaluation. |
|
Risk analysis |
The estimation of the risk associated with the identified hazards |
|
Risk evaluation |
The comparison of the estimated risk to given risk criteria, using a quantative or qualitative scale, to determine the significance of the risk |
|
Risk control |
contains risk reduction or risk acceptance |
|
Risk review |
contains an evaluation or review after a period of time to assess the measures taken and define if the identified risks are reduced effectiveley |
Important definitions for risk management process
I requisiti della norma ISO 14971: 2012 sono applicabili a tutte le fasi del ciclo di vita di un dispositivo medico.
La gestione dei rischi è il processo di gestione della qualità complessiva con cui i rischi sono identificati, valutati, controllati, monitorati e rivisti. Il rischio può essere stimato basandosi su quanto segue:
- - Gravità (Impact) = il grado di danno
- - Probabilità / evento / possibilità = il tasso probabile di occorrenza
Rischio = Probabilità di impatto x Gravità

L'analisi dei rischi è uno strumento importante che consente di ottimizzare il progetto, considerando i possibili rischi connessi con un nuovo prodotto. Risk Analysis è un documento che deve essere impostato nelle prime fasi di definizione del progetto.
In questo modo è possibile valutare le contromisure atte a ridurre il rischio.
L'analisi dei rischi deve dimostrare che i rischi sono stati valutati e ha agito per ridurre l'impatto.